Myocardial infarction is considered part of a spectrum referred to as acute coronary syndrome (ACS).
The ACS continuum representing on going myocardial ischemia or injury consists of unstable angina, non–ST-segment elevation myocardial infarction (NSTEMI), and ST-segment elevation myocardial infarction (STEMI).
Patients with ischemic discomfort may or may not have ST-segment or T-wave changes denoted on the electrocardiogram (ECG).
ST elevations seen on the ECG reflect active and ongoing transmural myocardial injury.
Without immediate reperfusion therapy, most persons with STEMI develop Q waves, reflecting a dead zone of myocardium that has undergone irreversible damage and death. Those without ST elevations are diagnosed either with unstable angina or NSTEMI―differentiated by the presence of cardiac enzymes.
Both these conditions may or may not have changes on the surface ECG, includingST-segmentdepression or T-wave morphological changes.
Patofisologi
The spectrum of myocardial injury --> impaired myocardial perfusion and metabolic demand at the time of the event.
The damage in the myocardium is essentially the result of a tissue response that includes apoptosis (cell death) and inflammatory changes. Therefore, the hearts of patients who suddenly die from an acute coronary event may show little or no evidence of damage response to the myocardium at autopsy.
The typical myocardial infarction initially manifests as coagulation necrosis that is ultimately followed by myocardial fibrosis. Contraction-band necrosis is also seen in many patients with ischemia. This is followed by reperfusion, or it is accompanied by massive adrenergic stimulation, often with concomitant myocytolysis.
Inferior-wall myocardial infarction and right ventricular myocardial infarction
In severe cases of acute inferior-wall myocardial infarction with RV involvement, the forward delivery of blood from the RV to the LV may be insufficient to fill the LV, resulting in low blood pressure even if the LV is intact. (See Physical Examination.)
Chemoreceptor activation in the myocardium actuates vagal (parasympathetic) efferent discharge, known as the Bezold-Jarisch reflex, which causes bradycardia and vessel dilation that may further lower blood pressure. Adenosine may accumulate in the infarct zone secondary to a local inhibition of adenosine deaminase, for which aminophylline may act pharmacologically as an antagonist. The hemodynamic changes resemble many of those seen with pericardial constriction or tamponade. Patients with this condition respond well to an infusion of normal sodium chloride solution. Improvement with such infusion compensates for failure of the pumping action of the RV; it reduces vagal tone, and it deactivates the pressure sensors that were sending a hormonal signal to the kidneys to retain salt.
Arrhythmogenesis
In addition to the direct effects of ischemia and tissue hypoxia, decreased removal of noxious metabolites, including potassium, calcium, amphophilic lipids, and oxygen-centered free radicals, also impair ventricular performance. These abnormalities promote potentially lethal arrhythmias.
Pericarditis
Epicardial inflammation may initiate pericarditis, which is seen in more than 20% of patients presenting with Q-wave infarctions.
Reduced systolic function
Lack of adequate oxygen and insufficient metabolite delivery to the myocardium diminish the force of muscular contraction and decrease systolic wall motion in the affected territory.
Abnormal regional wall motion
Even brief deprivation of oxygen and the requisite metabolites to the myocardium diminishes diastolic relaxation and causes abnormal regional systolic contractile function, wall thickening, and abnormal wall motion. If the area affected is extensive, diminished stroke volume and cardiac output may result.
Hypokinesis and akinesis
In general, regions of hypokinesis and akinesis of the ventricular myocardium reflect the location and extent of myocardial injury. Evidence of hypokinesis is seen on the echocardiogram below.
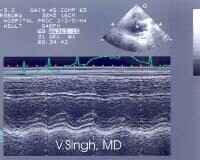 Hypokinesis of the anteroseptal wall observed during echocardiography in a patient presenting with an acute anteroseptal myocardial infarction.
Hypokinesis of the anteroseptal wall observed during echocardiography in a patient presenting with an acute anteroseptal myocardial infarction.
Myocardial infarction expansion
In general, expansion of infarcted myocardium and resultant ventricular dilatation (ie, ventricular remodeling) ensues within a few hours after the onset of a myocardial infarction. An expanding myocardial infarction leads to thinning of the infarct zone and realignment of layers of tissue in and adjacent to it, causing ventricular dilatation.
Myocardial rupture
Myocardial rupture was seen in as many as 10% of fatal myocardial infarctions before the era of thrombolytics, but it is now encountered much less often. When rupture occurs, it may be associated with large infarctions; indications include cardiogenic shock or hemodynamically significant arrhythmia. Patients may have a history of hypertension with ventricular hypertrophy.
Ventricular aneurysm
A ventricular aneurysm is an outward bulging of a noncontracting segment. In the early days of cardiac imaging, ventricular aneurysms were seen in as many as 20% of patients with Q-wave myocardial infarction, but now it is seen in less than 8%.
Cardiogenic shock
In patients with extensive myocardial injury, coronary blood flow diminishes as cardiac output declines and heart rate accelerates. Because coronary artery disease is usually generalized or diffuse, ischemia that occurs at a distance from the infracted segment may result in a vicious cycle in which a stuttering and expanding myocardial infarction ultimately leads to profound LV failure, hypotension, and cardiogenic shock.
Effect on diastolic function
Immediately after the onset of myocardial infarction, the ability of ischemic myocardium to relax declines. Relaxation is an active process that uses ATP. Impaired relaxation increases LV end-diastolic volume (LVEDV) and LV end-diastolic pressure (LVEDP).
The increased LVEDP results in ventricular dilation, increased pulmonary venous pressure, decreased pulmonary compliance, and interstitial and (ultimately) alveolar pulmonary edema. These effects lead to increased hypoxemia, which may worsen ischemic injury to the myocardium.
Etiology
Atherosclerosis is the disease primarily responsible for most acute coronary syndrome (ACS) cases. Approximately 90% of myocardial infarctions result from an acute thrombus that obstructs an atherosclerotic coronary artery. Plaque rupture and erosion are considered to be the major triggers for coronary thrombosis. Following plaque erosion or rupture, platelet activation and aggregation, coagulation pathway activation, and endothelial vasoconstriction occur, leading to coronary thrombosis and occlusion.
Within the coronary vasculature, flow dynamics and endothelial shear stress are implicated in the pathogenesis of vulnerable plaque formation.Evidence indicates that in numerous cases, culprit lesions are stenoses of less than 70% and are located proximally within the coronary tree.Coronary atherosclerosis is especially prominent near branching points of vessels.Culprit lesions that are particularly prone to rupture are atheromas containing abundant macrophages, a large lipid-rich core surrounded by a thinned fibrous cap.
Nonmodifiable risk factors for atherosclerosis include the following:
- Age
- Sex
- Family history of premature coronary heart disease
- Male-pattern baldness
Modifiable risk factors for atherosclerosis include the following:
- Smoking or other tobacco use
- Diabetes mellitus
- Hypertension
- Hypercholesterolemia and hypertriglyceridemia, including inherited lipoprotein disorders
- Dyslipidemia
- Obesity
- Sedentary lifestyle and/or lack of exercise
- Psychosocial stress
- Poor oral hygiene
- Type A personality
Elevated homocysteine levels and the presence of peripheral vascular disease are also risk factors for atherosclerosis.
Intramural thrombus development
Inflammation of the endocardial surfaces and stasis of blood flow associated with regional akinesis (no wall motion) or dyskinesis (abnormal, passively reversed wall motion) may lead to the formation of ventricular mural thrombi, which have the potential to embolize.
Patients with acute myocardial infarction are prone to cerebrovascular injury as a result of emboli from ventricular mural thrombi; the rate is approximately 1%.
Causes of myocardial infarction other than atherosclerosis
Nonatherosclerotic causes of myocardial infarction include the following:
- Coronary occlusion secondary to vasculitis
- Ventricular hypertrophy (eg, left ventricular hypertrophy, idiopathic hypertrophic subaortic stenosis [IHSS], underlying valve disease)
- Coronary artery emboli, secondary to cholesterol, air, or the products of sepsis
- Congenital coronary anomalies
- Coronary trauma
- Primary coronary vasospasm (variant angina)
- Drug use (eg, cocaine, amphetamines, ephedrine)
- Arteritis
- Coronary anomalies, including aneurysms of coronary arteries
- Factors that increase oxygen requirement, such as heavy exertion, fever, or hyperthyroidism
- Factors that decrease oxygen delivery, such as hypoxemia of severe anemia
- Aortic dissection, with retrograde involvement of the coronary arteries
- Infected cardiac valve through a patent foramen ovale (PFO)
- Significant gastrointestinal bleed
In addition, myocardial infarction can result from hypoxia due to carbon monoxide poisoning or acute pulmonary disorders. Infarcts due to pulmonary disease usually occur when demand on the myocardium dramatically increases relative to the available blood supply.
Although rare, pediatric coronary artery disease may be seen with Marfan syndrome, Kawasaki disease, Takayasu arteritis, progeria, and cystic medial necrosis.
Imaging studies, such as contrast chest CT scans or transesophageal echocardiograms, should be used to differentiate myocardial infarction from aortic dissection in patients in whom the diagnosis is in doubt. Stanford type A aortic dissections may dissect in a retrograde fashion causing coronary blockage and dissection, which may result in myocardial infarction. In one study, 8% of patients with Stanford type A dissections had ST elevation on ECG. (See Echocardiography.)
Myocardial infarction induced by chest trauma has also been reported, usually following severe chest trauma such as motor vehicle accidents and sports injuries.
Acute myocardial infarction in childhood
Acute myocardial infarction is rare in childhood and adolescence . Although adults acquire coronary artery disease from lifelong deposition of atheroma and plaque, which causes coronary artery spasm and thrombosis, children with acute myocardial infarction usually have either an acute inflammatory condition of the coronary arteries or an anomalous origin of the left coronary artery. Intrauterine myocardial infarction also does occur, often in association with coronary artery stenosis.
Prognosis
One third of patients who experience STEMI die within 24 hours of the onset of ischemia, and many of the survivors experience significant morbidity. However, a steady decline has occurred in the mortality rate from STEMI over the last several decades.
Acute myocardial infarction is associated with a 30% mortality rate; half of the deaths occur prior to arrival at the hospital. An additional 5-10% of survivors die within the first year after their myocardial infarction. Approximately half of all patients with a myocardial infarction are rehospitalized within 1 year of their index event.
In a study that assessed the impact of prehospital time on STEMI outcome, Chughatai et al suggest that "total time to treatment" should be used as a core measure instead of "door-to-balloon time."This is because on-scene time was the biggest fraction of "prehospital time." The study compared groups with total time to treatment of more than 120 minutes compared with 120 minutes or less and found mortalities were 4 compared with 0 and transfers to a tertiary care facility were 3 compared with 1, respectively.
Overall, prognosis is highly variable and depends largely on the extent of the infarct, the residual left ventricular function, and whether the patient underwent revascularization.
Better prognosis is associated with the following factors:
- Successful early reperfusion (STEMI goals: patient arrival to fibrinolysis infusion within 30 minutes OR patient arrival to percutaneous coronary intervention within 90 minutes)
- Preserved left ventricular function
- Short-term and long-term treatment with beta-blockers, aspirin, and ACE inhibitors
Poorer prognosis is associated with the following factors:
- Increasing age
- Diabetes
- Previous vascular disease (ie, cerebrovascular disease or peripheral vascular disease)
- Elevated Thrombolysis in Myocardial Infarction (TIMI) risk score for unstable angina/NSTEMI (7 factors: Age ≥65 y, ≥3 risk factors for cardiac disease, previous coronary disease, ST segment deviation ≥0.5 mm, ≥2 episodes of angina in last 24 h, aspirin use within prior wk, and elevated cardiac enzyme levels)
- Delayed or unsuccessful reperfusion
- Poorly preserved left ventricular function (the strongest predictor of outcome)
- Evidence of congestive heart failure (Killip classification ≥II)or frank pulmonary edema (Killip classification ≥III)
- Elevated B-type natriuretic peptide (BNP) levels
- Elevated high sensitive C-reactive protein (hs-CRP), a nonspecific inflammatory marker
- Secretory-associated phospholipase A2 activity is related to atherosclerosis and predicts all-cause mortality in elderly patients; it also predicts mortality or MI in post-MI patients
Blood glucose
Beck et al found that elevated blood glucose level on admission is associated with increased short-term mortality in nondiabetic patients presenting with a first acute myocardial infarction. Analysis of data from a German myocardial infarction registry database showed that among 1,631 nondiabetic acute myocardial infarction patients with admission glucose level more than 152 mg/dL (top quartile), the risk of death within 28 days was higher than among patients in the bottom quartile (odds ratio, 2.82; 95% confidence interval, 1.30-6.12). However, in 659 registry patients with type 2 diabetes, admission glucose levels did not correlate significantly with short-term mortality. Beck et al concluded that nondiabetic acute myocardial infarction patients with elevated glucose levels constitute a high-risk group that requires aggressive intervention.[22]
Psychological depression
The combination of acute myocardial infarction and psychological depression appears to worsen the patient's prognosis. Acute myocardial infarction may precipitate reactive depression whether or not beta-adrenergic blocking agents or other CNS-active agents are administered.
Myocardial hibernation and stunning
After the occurrence of 1 or more ischemic insults, impaired wall motion is often transient (myocardial stunning) or prolonged (myocardial hibernation). These phenomena occur because of the loss of essential metabolites such as adenosine, which is needed for adenosine triphosphate (ATP)–dependent contraction. Hibernation, a persisting wall-motion abnormality that is curable with revascularization, must be differentiated from permanent, irreversible damage or completed infarct.
Scar tissue and prognosis
Scars involving less than one third of the thickness of the wall, as shown on contrast-enhanced MRI, likely correspond to a recovery of myocardial function, whereas with scars measuring more than one third the thickness of the wall, the potential for recovery with therapy is limited (except in cases involving research cell therapies or surgical scar revision). Other findings associated with recovery are activity on 2-[Fluorine 18]-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography (PET) scanning and a monophasic or biphasic contractile response to dobutamine infusion, caused by the induction of ischemia. Cardiac scar tissue is seen in the image below.
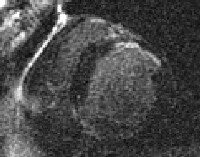 Image shows a scar in the anterior wall.
Image shows a scar in the anterior wall.
No comments:
Post a Comment